
A information supplied by the manufacturer to inform the user of a product’s intended purpose and appropriate use is referred to as “instructions for use.” The following items must be present in the information provided with the directions for use:
- Information in line with Section 23.2 of the MDR points (a), (c), (e), (f), (k), (l), (n), and (r);
- A thorough description of the device’s intended use
- The product’s performance features
- A description of the predicted clinical advantages, if applicable
- a list of any residual risks, contraindications, or unfavourable side effects
- Information required to ensure the device’s correct installation and operation
- In the event that sterile packing is broken, instructions for handling sterile equipment are provided
- Instructions for sterilization of provided non-sterile equipment, where applicable
- The user manual’s publication date ( revision number, document ID also is important)
- If the equipment emits radiation for medicinal purposes, this information should be included
- Any additional criteria for the device user’s facilities or qualifications
- Cleaning, disinfecting, and packing of reusable equipment are examples of acceptable methods for permitting re-use
- The total qualitative and quantitative information on the materials to which patients may be exposed in the case of implanted goods
- Information on known features and technical elements that may cause a risk if the product were to be re-used in the case of single-use devices
- Any precautions or warnings that will aid in the safe disposal of the medical equipment, its attachments, and any consumables
- The situations in which a layperson should seek the advice of a healthcare expert.
EU MDR Requirements for User Manual / IFU as per the below-harmonized standard must be followed by all manufacturers.
EN ISO 15223-1:2016 Medical devices – Symbols to be used with medical device labels, labelling and information to be supplied – Part 1: General requirements (ISO 15223-1:2016, Corrected version 2017-03)
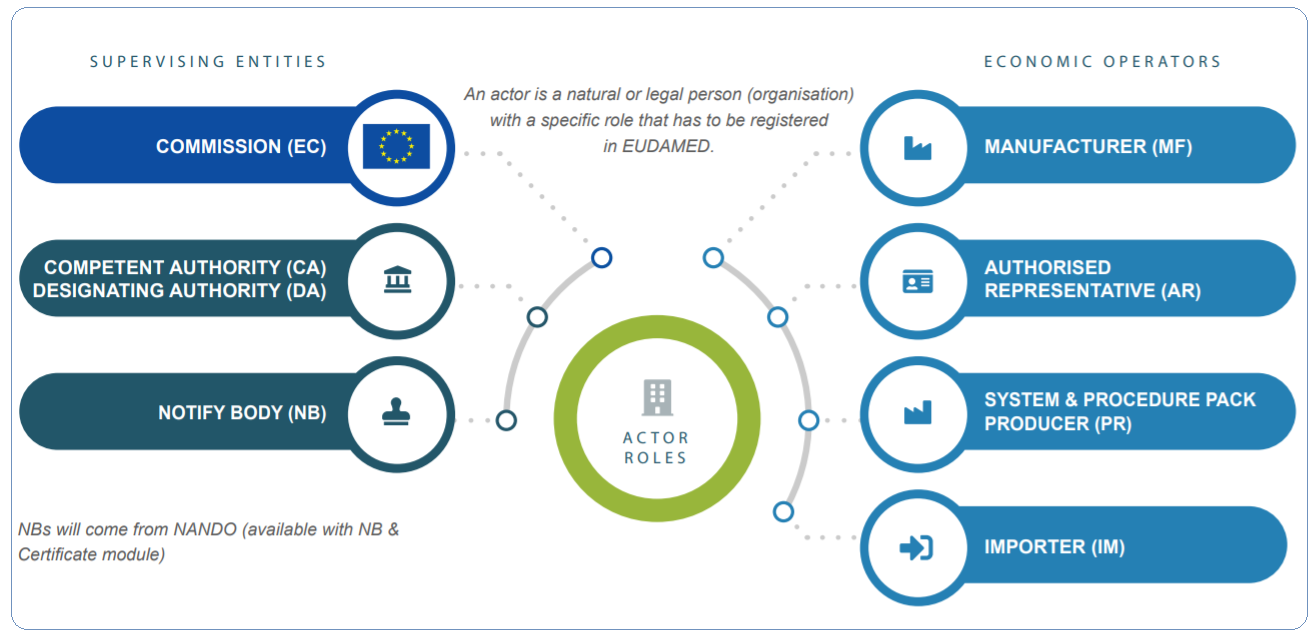
The MDR also applies to medical devices and accessories used in clinical trials, as well as the product groupings without a medical purpose mentioned in Annex XVI (e.g., contact lenses, liposuction equipment, dermatology equipment).
The following products fall outside of the scope of the MDR:
- Medical diagnostic devices used in vitro analysis
- Directive 2001/83/EC applies to medical goods
- Advanced treatment medical goods are covered under Regulation (EC) No 1394/2007
- Human blood, blood cells, plasma, or other blood products are all examples of blood products.
Failure to follow correct labelling regulations might result in Notified Body objections, unnecessary compliance problems, EU port of entry holds, and costly product recalls. As a result, manufacturers must be familiar with the new EU MDR labelling requirements.
- What percentage of your device labelling is effective?
- Are they in accordance with the specifications?
- Consult with an expert consultant for a clear review, and keep your label/IFU/User Manual current to ensure compliance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}